
3 min read
Biopharmaceutical products represent up to 20 percent of the total pharmaceutical market and are growing at a rate of nearly eight percent annually. To keep up with this impressive growth rate, many contract development and manufacturing organizations (CDMOs) are making strategic investments in equipment and expertise to support analytical development and structural characterization of biopharmaceuticals in more economical and efficient ways.[1]
Unlike small molecule active pharmaceutical ingredients (APIs) that exist as a single chemical entity, biologics nearly always exist as a mixture of molecules. Different molecules can arise from numerous sources including N-terminal variants, post-translation modifications (PTMs), glycoforms, and degradation products. Due to the potentially high amount of heterogeneity in the biologic, it is critical to demonstrate control over the drug substance (DS) manufacturing process from fermentation to purification and protein re-folding. Characterization of the various molecules is the first step in demonstrating process control. High-resolution mass spectrometry is a key component in the characterization of novel biologics and biosimilars.
INTACT MASS ANALYSIS
Intact mass analysis of biologics is an important tool for verifying that the purified drug substance was successfully expressed and purified. Intact mass analysis of monoclonal antibodies (mAbs) verifies that the molecule has been assembled correctly and all expected post-translation modifications are present. While this provides a high-level confirmation of protein molecular weight, it cannot provide the location
of any modifications that might be present. Intact mass analysis is the first step in the structural characterization of biologics.
PEPTIDE MAPPING
Further characterization of biologics requires the analysis of smaller segments of the protein. Peptide mapping is one technique that can provide the necessary resolution to ensure that desired modifications are present, and specific impurities are controlled where traditional chromatographic or electrophoretic techniques prove to be difficult. Coupled with high-resolution mass spectrometry, peptide mapping can provide acceptable specificity, repeatability, and accuracy to quantitate known degradation peptides within the digested sample.
Peptide mapping generally consists of the following steps:
- Denaturation of the target protein
- Reduction of disulfide bonds
- Alkylation of the free thiol groups within the side chains of cysteine residues to ensure disulfide bonds are not reformed
- Buffer exchange into digestion buffer
- Digestion with an appropriate endoproteinase
- Analysis by HPLC/UV/MS or MS/MS
Glycoproteins can be deglycosylated prior to performing peptide mapping to reduce the heterogeneity introduced by various glycoforms. Identification of peptides generated during the peptide mapping procedure can be attained by using advanced mass spectrometry software tools based on intact mass and MS/MS fragmentation patterns of the peptides.
The use of peptide mapping to characterize biologics provides the advantage of the protein being divided into a large number of smaller peptides. The generated peptides can then be chromatographically separated using HPLC or UPLC technology. The intact masses of the peptides can provide information regarding the type of modification that was made. MS/MS sequencing of the peptides can indicate the exact amino acid residue containing the modification. This level of resolution at the individual amino acid level is simply not possible with mass spectrometric analysis of intact proteins or mAbs. Examples of the use of peptide mapping to characterize and quantitate specific posttranslation modifications or degradation products are provided.
N-TERMINAL VARIANTS
Quantitation of N-terminal variants from bacterial (formyl-Met, Met, or another amino acid) expression systems, eukaryotic (i.e., pyro-Glu, Gln) expression systems, or synthetic peptides (acetylation) are possible using the correctly chosen endoproteinase for digestion. Chromatographic separation of peptides that differ by a single amino acid is generally easier than separating intact proteins that only differ by a single amino acid. Given that this type of peptide mapping is targeting a single peptide from the entire protein, the chromatography can be optimized for the N-terminal peptide and its variants rather than optimized to separate all peptides generated from the peptide map. Due to the relatively large mass differences between the different N-terminal variants, quantitation can be performed using multiple reaction monitoring (MRM) detection on triple-quadrupole mass spectrometers, allowing for maximum specificity and adequate sensitivity to observe even trace levels of minor components.
DEGRADATION PRODUCTS
Quantitation of degradation products generated during protein purification and refolding steps or during stability assessment of the drug product, such as methionine oxidation or deamidation of asparagine and glutamine residues, can be more challenging given the potentially larger number of degradation locations and their distribution in the protein. Oxidation of methionine residues is one of the most common degradation products found in biologics. Methionine can be oxidized through oxygen dissolved in the buffer, or through the formation of hydroxyl radicals upon exposure to UV light. Deamidation of asparagine residues can occur during protein purification and refolding steps or during the shelf-life of the drug product. Deamidation occurs faster at higher pH and higher temperature and is more likely for asparagine than it is for glutamine. The amino acid following the asparagine or glutamine residue can also affect the rate of deamidation.
Digestions should be performed with the goal of generating the largest number of peptides that contain only a single potential modification site. In the situation where a single endoproteinase will not generate an acceptable peptide map for the characterization and quantitation of degradation products, sequential or simultaneous digestions with multiple endoproteinases may need to be performed. Given the small mass difference between asparagine and aspartate residues that result from deamidation, quantitation is best done using high-resolution mass spectrometry. Scanning a mass range can allow for the detection of multiple degradation products with a single injection using extracted ion chromatograms (XICs) of the native and modified peptides.
Peptide mapping with high-resolution mass spectrometry can provide both the peptide-specific location of the modification and the ability to observe small mass changes.
DISULFIDE BOND FORMATION
Correct disulfide bond formation can be critical to protein folding, enzyme activity, and proper binding in biologics. Formation of incorrect intra-molecular disulfide bonds can lead to misfolded proteins and reduced activity or binding. Formation of incorrect inter-molecular disulfide bonds can also lead to protein aggregation, which is of great immunogenicity concern to patients. A schematic of a typical mAb
showing intra-molecular and inter-molecular disulfide bonds is provided in Figure 1.
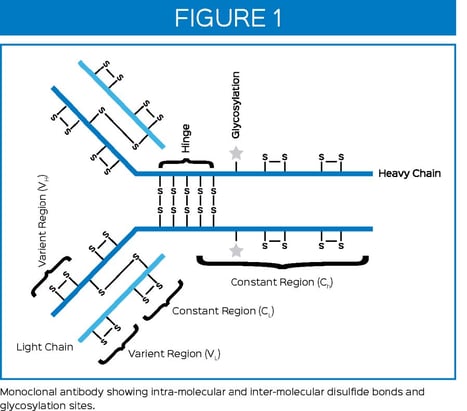
During peptide mapping, disulfide bonds are typically reduced (using DTT, TCEP, or ß-ME) then alkylated (using Iodoacetamide or Iodoacetic acid) to prevent reformation of disulfide bonds during subsequent steps. Therefore, information regarding which disulfide bonds were present in the protein prior to peptide mapping is lost. However, non-reducing peptide maps can be performed, allowing information regarding which disulfide bonds are present to be retained. The non-reducing peptide map should be performed in parallel with the traditional peptide map reduction and analyzed using the same HPLC or UPLC method with MS detection. Peptides that are involved in disulfide bonds will have peaks with different retention times in the non-reduced peptide map from the retention times of the corresponding individual peptide peaks in the reduced peptide map. Comparison of the chromatograms between the reduced and non-reduced peptide maps should quickly reveal which peaks were involved in the disulfide bonds. A high-resolution mass spectrometer can then determine which peptides are linked through disulfide bonds using intact mass.
CONFIRMATION OF PROTEIN SEQUENCE BY MS/MS
In order to gain specific information from amino acid residue from peptide mapping, sequencing of peptides by tandem mass spectrometry (MS/MS) is required. Peptides are sequenced using collisioninduced dissociation (CID) to generate product ions from a single or set of precursor ions. Peptides will fragment in predictable ways along the peptide backbone to yield a series of product ions (Figure 2). The a, b, and c-series product ions provide information toward the N-terminal side of the peptide while the x, y, z-series ions provide information towards the C-terminal side of the peptide. The b-series and yseries ions can be used to sequence the peptide, confirming the presence of each amino acid.
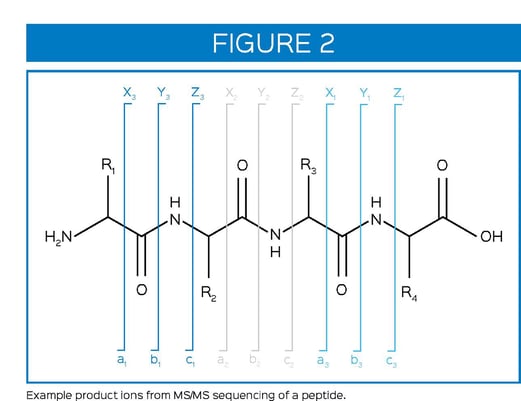
MS/MS sequencing can be used to determine the residue-specific location of a post-translation modification or degradation product. It can also be used as an orthogonal technique to N-terminal sequencing using Edman degradation. Advantages over Edman degradation are 1) MS/MS sequencing works even if the N-terminus is blocked (eg, acetylated, pyro-Glu) and 2) can provide sequencing information for most of a protein, while Edman degradation is typically limited to less than 40 residues. MS/MS sequencing can even be used to identify unknown proteins by comparing peptide sequences to a protein library database.
GLYCOFORMS
One of the most complex post-translational modifications is glycosylation of proteins and mAbs. There are dozens of glycans ranging from high mannose glycans to hybrid glycans to complex glycans. Glycosylation can be important for protein function but may also play a role in immunogenicity. N-linked glycans are attached to the side-chain of an asparagine residue within the sequence Asn-X-Ser or Asn-X
Thr where X is any amino acid except proline. O-linked glycans are attached to the side chains of serine and threonine residues and there is no known consensus sequence.
Monoclonal antibodies contain a single N-linked glycosylation site in each heavy chain while other proteins may contain multiple glycosylation sites. Glycan profiles are largely determined by culture conditions and the genotypes of host cells. Characterization of glycosylation is a critical step to demonstrating control over the bioprocess used to express proteins and mAbs.
Most N-linked glycans can be removed using PNGase F. The released glycans may be analyzed by MALDITOF, or they can be labeled with a fluorescent tag and analyzed by hydrophilic interaction chromatography (HILIC) with mass spectrometry detection. Labeling of glycans with a fluorescent tag has several advantages. The labeled glycans can be chromatographically separated using HILIC and quantitated using highly sensitive fluorescence detection. The HILIC method should be compatible with mass spectrometry, allowing for the characterization of the labeled glycan by intact mass. Product ions resulting from the in-source fragmentation of glycosidic bonds are often observed.
Characterization of the glycosylation site within the protein can be performed using the aforementioned peptide mapping procedures. The glycosylated peptide should contain multiple masses corresponding to the various glycoforms. Deglycosylation of the protein prior to peptide mapping should result in a single peptide mass in place of the multiple masses observed for the glycosylated peptide. It should be noted that glycosylation may not change the retention time of the peptide as retention on a reversed-phase HPLC column is driven by hydrophobic interactions, to which the glycans contribute very little. The peptide map can also be analyzed using HILIC. As the glycans are well retained in this separation technique, the glycopeptides are well separated from the non-glycosylated peptides.
SUMMARY
High-resolution mass spectrometry is a core technique for the characterization of biologics. It can provide a full range of characterization capabilities ranging from high-level analysis through intact mass analysis to residue specific information from MS/MS sequencing.
Alcami offers a full range of capabilities for the structural characterization of biologics using high resolution mass spectrometry. Instrumentation includes two high-resolution quadruple time of flight (Qtof) instruments and multiple high-sensitivity triple-quadrupole instruments.
Q-Tof Mass Spectrometers
- Intact mass analysis
- Identification of peptides from peptide maps
- Confirmation of disulfide bond formation
- Identification of unknown impurities including methionine oxidation and deamidation
- Analysis of glycoforms
- MS/MS sequencing
Triple-Quadrupole Mass Spectrometers
- Quantitation of specific impurities
- Quantitation of N-terminal variants
ABOUT THE AUTHOR
Dr. William Boomershine is a Principal Scientist at Alcami Corporation. He earned his Ph.D. in Biochemistry from The Ohio State University for his research on the solution structures of archaeal Ribonuclease P proteins using NMR. He has more than 12 years of experience in the pharmaceutical development and manufacturing industry characterizing small molecule APIs, peptides, and proteins using high-resolution mass spectrometry. He possesses a strong knowledge base in peptide and protein chemistry and development of analytical methods for peptides and proteins.
[1] McKinsey & Company 2014. Rapid growth in biopharma: challenges and opportunities.
Website visited: https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/rapid-growth-in-biopharma.
7 min read
White Paper: Host Cell Impurities, Bioassays, Micro-Flow Imaging
As opposed to traditional drug production, the production of biological products (biologics) requires the involvement of human, animal, or microbial...